Journal of
Clinical & Medical Surgery
Clinical & Medical Surgery
www.jclinmedsurgery.com
ISSN 2833-5465
Open Access
Volume 5
Open Access
Volume 5
Armaan Siddiqui1; Ayan Purwar1; Rahul Purwar2; Sushant Kumar1*
*Corresponding Author: Sushant Kumar
Senior Scientist, Immuno ACT, Immunoadoptive Cell Therapy Private Limited (ImmunoACT), India.
Email: sushant.kumar@immunoact.com
Article Info
Received: Aug 28, 2025
Accepted: Sep 30, 2025
Published: Oct 07, 2025
Archived: www.jclinmedsurgery.com
Copyright: © Kumar S (2025).
Abstract...
Chimeric Antigen Receptor-T cell (CAR-T) therapy has transformed the treatment of hematologic malignancies, achieving durable remissions in patients with refractory leukemia, lymphoma, and myeloma. However, the current ex vivo manufacturing method is resource-intensive, time-consuming, and very expensive, limiting global accessibility. The multi-step process includes leukapheresis, genetic modification, expansion, and stringent quality control under Good Manufacturing Practice (GMP) conditions. These complexities contribute to high production costs, long vein-to-vein times, and manufacturing failures in heavily pretreated patients. These challenges underscore the need for innovative strategies to broaden the reach of this transformative modality. In vivo engineering of CAR-T cells offers a promising solution by directly delivering genetic material into circulating T cells, bypassing ex vivo manipulation. Among nonviral vectors, Lipid Nanoparticles (LNPs) have emerged as the leading platform due to their clinical validation in nucleic acid delivery, scalability, and favorable safety profile. Advances in ionizable lipid chemistry, helper lipid selection, and Polyethylene Glycol (PEG)-lipid optimization have enabled efficient nucleic acid encapsulation, targeted delivery, and controlled release. Furthermore, selective organ targeting approaches and antibody conjugation techniques have enhanced T-cell specificity, while transient mRNA-based delivery mitigates integration-related risks. Preclinical studies in murine and nonhuman primate models demonstrate that targeted LNPs can generate functional CAR-T cells in vivo, achieve tumor clearance, and exhibit manageable toxicity profiles. This review outlines the evolution of CAR-T therapy, critically examines the limitations of current ex vivo approaches, and highlights recent advances in LNP-based in vivo CAR-T engineering. We discuss design principles, preclinical progress, safety considerations, and translational prospects of this approach. If clinical efficacy and safety are confirmed, LNP-mediated in vivo CAR-T generation has the potential to redefine cancer immunotherapy by reducing costs, shortening treatment timelines, and expanding access to patients worldwide.
Keywords: CAR-T; Lipid nanoparticles; In vivo engineering; Gene delivery; Cancer immunotherapy.
Abbrevations: aCD3-LNP: Anti-CD3-conjugated Lipid Nanoparticle; aLNP: Activating Lipid Nanoparticle; ALL: Acute Lymphoblastic Leukemia; CAR: Chimeric Antigen Receptor; CAR-T: Chimeric Antigen Receptor T-cell; CLL: Chronic Lymphocytic Leukemia; CRS: Cytokine Release Syndrome; CTL: Cytotoxic T Lymphocyte (if mentioned implicitly in context of T-cell responses); DLS: Dynamic Light Scattering; DSPC: Distearoylphosphatidylcholine; DSPE-PEG: 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-polyethylene glycol; DOPE: Dioleoylphosphatidylethanolamine; FDA: Food and Drug Administration; Fc: Fragment Crystallizable (region of antibody); Fluc: Firefly Luciferase (reporter gene); GMP: Good Manufacturing Practice; ICANS: Immune Effector Cell–Associated Neurotoxicity Syndrome; IL-6: Interleukin 6; IND: Investigational New Drug; LNP: Lipid Nanoparticle; mRNA: Messenger RNA; MM: Multiple Myeloma; NHL: Non-Hodgkin Lymphoma; NSG: NOD-scid IL2Rγnull (immunodeficient mouse strain); oRNA: Circular RNA (Orna Therapeutics platform); PBAE: Poly (β-amino ester); PBMC: Peripheral Blood Mononuclear Cell; PDI: Polydispersity Index; PEG: Polyethylene Glycol; scFv: Single-chain Variable Fragment; SORT: Selective Organ Targeting; STING: Stimulator of Interferon Genes; tLNP: Targeted Lipid Nanoparticle; TRUCK: T cells Redirected for Universal Cytokine Killing.
Citation: Siddiqui A, Purwar A, Purwar R, Kumar S. Next-generation CAR-T: In vivo engineering with lipid nanoparticles. J Clin Med Surgery. 2025; 5(2): 1208.
Introduction
Chimeric Antigen Receptor-T cell (CAR-T) therapy has emerged as a transformative strategy in oncology, demonstrating extraordinary efficacy in hematologic malignancies [1]. Durable remissions have been achieved in patients with Acute Lymphoblastic Leukemia (ALL), Non-Hodgkin Lymphoma (NHL), Chronic Lymphocytic Leukemia (CLL), and Multiple Myeloma (MM), many of whom had exhausted conventional treatment options [1,2]. To date, seven CAR-T products have received US FDA approval, including tisagenlecleucel (Kymriah), axicabtagene ciloleucel (Yescarta), brexucabtagene autoleucel (Tecartus), lisocabtagene maraleucel (Breyanzi), idecabtagene vicleucel (Abecma) and ciltacabtagene autoleucel (Carvykti). Recently, Talicabtagene autoleucel (NexCAR19), a CD19-directed CAR-T therapy developed in India, has been shown to improve affordability and accessibility [4-6].
Despite these advances, broad clinical implementation is limited by challenges intrinsic to the ex vivo manufacturing process [7]. The cost of commercial CAR-T products frequently exceeds USD 373,000, restricting access even in high-income countries and rendering them inaccessible in most resource limited settings [8]. Production requires leukapheresis, genetic modification, expansion, and quality control under Good Manufacturing Practice (GMP) conditions, each step contributing to high resource demand and financial burden [9-12]. Logistical barriers further complicate delivery, with vein-to-vein times extending from weeks to months, a critical concern for patients with rapidly progressive disease [13]. Moreover, 4-7% of patients fail to receive therapy due to manufacturing failure or compromised T-cell fitness following extensive prior treatment, while sterility testing alone requires an additional 10-15 days [3,13-15].
To overcome these barriers, in vivo engineering of CAR-T cells is being explored as a next-generation approach. By delivering genetic material directly into patient T lymphocytes, this strategy aims to bypass ex vivo manipulation, reduce cost, and accelerate treatment availability. Preclinical studies employing viral and non-viral vectors, including lentiviruses, adenoassociated viruses, Lipid Nanoparticles (LNPs), and polymeric carriers, have demonstrated feasibility, underscoring the potential of in vivo CAR-T generation to expand access and scalability of this therapeutic modality [16]. However, viral vectors present additional limitations, including high manufacturing cost, strict biosafety requirements, and packaging size constraints, which have directed growing interest toward non-viral alternatives [17].
Among non-viral delivery systems, several cationic polymers have been extensively studied in preclinical settings. Polyethylenimine (PEI) is one of the most widely used, owing to its strong nucleic acid condensation capacity and efficient endosomal escape via the “proton sponge effect.” However, its clinical application is limited by significant cytotoxicity, particularly at higher molecular weights, and its non-degradability within cells. Poly (β-Amino Ester) (PBAE) polymers, in contrast, offer a biodegradable and biocompatible backbone with reduced toxicity, but suffer from low colloidal stability [18]. Chitosan, a naturally derived cationic polysaccharide, demonstrates biocompatibility, but its limited solubility at physiological pH restricts efficient systemic use [19]. Collectively, these platforms underscore the potential of polymer-based systems but have not advanced meaningfully into clinical use for CAR-T engineering.
In contrast, Lipid Nanoparticles (LNPs) have rapidly established as the most clinically validated non-viral delivery technology. Their success has been exemplified by the FDA approval of the siRNA drug Onpattro (patisiran) [20] and the global deployment of mRNA vaccines against COVID-19 [21], both of which highlighted their scalability, reproducibility, and safety profile. LNPs possess unique advantages, including efficient encapsulation of large nucleic acids, protection from nuclease degradation, delivery into different types of cells, and optimized endosomal escape through ionizable lipid components [20]. Furthermore, advances in Selective Organ Targeting (SORT) strategies now allow tailoring of biodistribution beyond the liver, broadening their potential utility in immune cell targeting [25]. Importantly, methods such as microfluidic mixing enable reproducible and scalable manufacturing, a critical factor for clinical translation [26]. Taken together, these attributes position LNPs as the leading non-viral platform currently under investigation for in vivo CAR-T generation. Early preclinical studies have demonstrated the ability of LNPs to deliver CAR-encoding mRNA or DNA directly to T cells in vivo and ongoing research is focused on improving targeting specificity, safety, and persistence. Thus, while polymeric systems have provided valuable mechanistic insights and proof-of-concept studies, LNPs represent the most advanced technology moving toward clinical translation in this space.
In this review, we summarize the evolution of CAR-T therapy, and the barriers associated with current ex vivo approaches, highlight recent advances in non-viral delivery systems with a focus on LNPs, and discuss their potential in enabling scalable, accessible, and clinically translatable in vivo CAR-T generation.
Evolution of chimeric antigen receptors
The fundamental design of Chimeric Antigen Receptors (CARs) has undergone iterative refinements since their initial conception, with each generation aimed at enhancing antitumor efficacy, persistence, and safety. (Figure 1) shows a canonical CAR structure, composed of an extracellular antigen-recognition domain, usually a single-chain variable fragment (scFv) derived from antibodies, a hinge and transmembrane domain, and intracellular signaling domains [27]. First-generation CARs contained only the CD3ζ signaling motif, which mediated cytotoxic activity but conferred limited persistence in vivo. Second generation CARs introduced costimulatory elements such as CD28 or 4-1BB, which markedly improved proliferation, survival, and durable responses, and remain the backbone of currently approved products [28]. Third-generation CARs integrated two costimulatory motifs in tandem, though their superiority over second-generation designs has not been consistently demonstrated in clinical settings [29]. More recently, fourth-generation CARs, often referred to as “TRUCKs” (T cells redirected for antigen-unrestricted cytokine-initiated killing), incorporate additional genetic modules to enable controlled cytokine release, resistance to exhaustion, or enhanced tumor trafficking [30]. These developments illustrate how molecular engineering of the CAR itself has been central to extending therapeutic impact, while also underscoring the complexity of optimizing receptor design in tandem with manufacturing feasibility.
Clinically, CAR-T therapies have demonstrated remarkable potential, inducing high remission rates in patients with hematologic malignancies that are otherwise treatment-resistant. Despite these successes, translating such outcomes into broad access remains challenging. The technical demands of producing CAR-T cells outside the body are significant, and the quality of T cells from heavily pretreated patients can be inconsistent, which sometimes limits both manufacturing success and therapeutic effectiveness [14]. Moreover, because each CAR-T product is patient-specific, scaling these therapies is inherently difficult; every batch is essentially a custom treatment rather than a standard, reproducible drug [31]. These challenges have underscored the need for next-generation approaches that can bypass the bottlenecks of ex vivo culture and individualized production, making these therapies more widely available.
The idea of generating CAR-T cells directly in vivo represents a significant shift in how these therapies could be delivered. Rather than removing, modifying, and reinfusing T cells, this approach aims to introduce genetic material straight into the patient’s circulating lymphocytes. If successful in the clinic, it could eliminate the need for leukapheresis, lengthy cell expansion, and extensive quality control, potentially reducing costs and shortening treatment time [32,33]. Early preclinical studies suggest that viral vectors, including lentiviruses and adeno-associated viruses, can transduce T cells in vivo [34-36]. However, these platforms face challenges such
as limited packaging capacity, biosafety concerns, and high manufacturing costs [37-41], prompting growing interest in non-viral alternatives that could offer lower immunogenicity, more flexibility in cargo size, and easier scale-up [42].
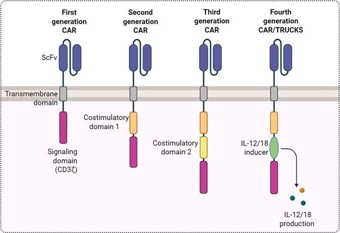
Figure 1: Fundamental design and different generations of Chimeric Antigen Receptors (CAR).
LNP design and components for In vivo CAR-T engineering
Among non-viral approaches, Lipid Nanoparticles (LNPs) have emerged as the most clinically advanced and translationally validated platform [24]. The development of Lipid Nanoparticles (LNPs) as non-viral vectors has opened new possibilities for directly generating CAR-T cells in vivo. Their design is modular, with each component exerting distinct effects on biodistribution, cellular uptake, and transfection efficiency. A typical LNP formulation includes ionizable lipids, helper lipids, cholesterol, and Polyethylene Glycol (PEG)-lipids (Figure 2), and optimization of each element is critical for efficient targeted delivery to T cells.
Ionizable lipids constitute the functional backbone of LNPs, conferring nucleic acid encapsulation, endosomal escape, and cytoplasmic release. Studies have identified specific ionizable lipids, like C14-4, L829, and DLINMC3DMA, as highly effective for T-cell engineering [43-46]. Their pKa is engineered to remain neutral in circulation, minimizing nonspecific interactions, and to become protonated in the acidic environment of endosomes. This facilitates membrane destabilization and promotes nucleic acid release into the cytosol [47,48]. Small modifications to ionizable lipid structure profoundly influence circulation half-life, tissue tropism, and ultimately the balance between efficacy and toxicity in T-cell [26,48].
Helper lipids such as Dioleoylphosphatidylethanolamine (DOPE) or Distearoylphosphatidylcholine (DSPC), stabilize LNP structure and enhance membrane fusion. Helper lipids play a central role in shaping lipid nanoparticle (LNP) biodistribution and, immunological outcomes, and confer distinct tropism profiles [48]. The fusogenic, unsaturated architecture of DOPE promotes a more fluid bilayer, facilitating endosomal escape and favoring hepatic delivery, with efficient mRNA uptake by hepatocytes and macrophages [49,50]. By contrast, DSPC, characterized by its saturated, rigid structure, drives LNP accumulation in the spleen [51,52]. Neither lipid directly activates T cells; their contribution is largely indirect through enabling mRNA delivery. Importantly, DOPE’s influence appears concentration dependent, while certain formulations enhance cytokine production, further increases in DOPE content do not consistently augment IL-2 secretion or T-cell proliferation [47].
PEG-lipids are incorporated at low molar ratios to reduce aggregation and extend circulation time by preventing opsonization [53]. The choice of PEG-lipid tail length and dissociation kinetics influences clearance rate and biodistribution, balancing the need for circulation stability with eventual cellular uptake [54]. DMG-PEG, with shorter C14 chains, sheds rapidly from the LNP surface, enabling faster cellular uptake, reduced anti-PEG antibody induction, and in some cases, improved lymph node delivery [ 55]. Conversely, DSPE-PEG (C18) integrates more stably, prolonging circulation but delaying endosomal escape and heightening anti-PEG IgM responses, which may trigger accelerated clearance upon repeat dosing [ 53]. For antibody conjugation, DSPE-PEG modified with maleimide is generally preferred due to its stable membrane anchoring, which supports durable ligand display, whereas DMG-PEG’s transient nature may compromise conjugate retention [53,56].
Cholesterol provides structural support and improves bilayer fluidity, impacting particle stability in circulation. Subtle variations in cholesterol analogs and percentage have been shown to affect LNP biodistribution and transfection efficiency, reflecting their contribution to system performance [57-60].
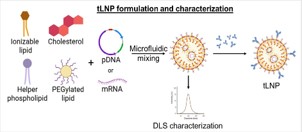
Figure 2: T-cell-targeted lipid nanoparticles (tLNPs) formulation.
Surface modification with antibody conjugates represents a particularly powerful strategy for directing LNPs toward T lymphocytes. Antibodies recognizing CD3, CD4, CD5 or CD8 [61] have been employed to restrict nucleic acid delivery specifically to T-cell subsets Beyond structural optimization, the choice of nucleic acid cargo remains central. Plasmid DNA can provide more durable CAR expression but require nuclear entry, resulting in lower transfection efficiency in primary T cells [62]. Messenger RNA (mRNA) has emerged as the dominant format for in vivo CAR-T engineering owing to its transient expression, absence of integration risk, and rapid translation into functional receptor proteins [63]. Collectively, these engineering principles form the basis of LNP design for in vivo CAR-T generation and continue to be refined through iterative preclinical experimentation.
Preclinical progress with LNP-mediated CAR-T engineering
Recent advances in Lipid Nanoparticle (LNP)-mediated in vivo CAR-T generation have demonstrated diverse strategies to achieve selective T-cell transfection, functional CAR expression, and therapeutic efficacy. Kheirolomoom et al. investigated whether anti-CD3-conjugated LNPs (aCD3-LNPs) could deliver reporter mRNA (mCherry or Fluc) specifically to T cells in situ to inform strategies for CAR-T generation. The LNPs contained the ionizable lipid DLin-MC3-DMA, DSPC, DSPE-PEG2k, DSPEPEG5k-Mal, DSPE-PEG2k-Cy7, and cholesterol, and were conjugated to a CD3ϵ F(ab’)2 fragment to minimize Fc-mediated effects. Systemic administration of 16% conjugated aCD3-LNPs in C57BL/6 mice transiently depleted splenic and circulating CD4+/ CD8a+ T cells while reducing CD3 surface expression. Circulating T cells recovered within 24 hours, and splenic T cells were activated, inducing Th1/Th2 cytokines and myeloid-derived chemokines. mCherry+ T cells reached in the spleen and in blood, with reporter-positive cells localizing to tumors and tumor-draining lymph nodes [44].
Zhou et al. aimed to simultaneously generate CAR-T cells and mitigate cytokine release by delivering CD19-CAR and IL-6 shRNA plasmids via CD3-modified LNPs. Nanoparticles (~200 nm, PDI <0.2, zeta potential ~0 mV) were intravenously injected at 5 µg/g in NSG mice following T-cell transfer. Stable CAR-T production persisted for 90 days, predominantly in CD8+ T cells, with peak expansion on day 21. IL-6 knockdown reduced cytokine release syndrome, and tumor burden decreased from day 12, achieving anti-tumor efficacy comparable to ex vivo CAR-T therapy [64].
Billingsley et al. explored antibody-functionalized C14-4 ionizable LNPs modified with maleimide-PEG for extrahepatic targeting. LNPs conjugated to antibodies against CD3, CD5, or CD7 delivered CD19 CAR mRNA intravenously at 0.5-2 mg/ kg. CD3-LNPs transfected 15-17% of circulating T cells at 12 hours, declining to 7% at 36 hours, while CD7-LNPs achieved 5-6% CAR positivity. Functional CAR-T cells depleted circulating B cells, with transient dose-dependent cytokine elevations. Untargeted LNPs produced negligible CAR expression, emphasizing the necessity of antibody-mediated targeting [65].
Hunter et al. evaluated targeted lipid nanoparticles (tLNPs) for direct CAR T cell engineering in mice and cynomolgus monkeys. In murine leukemia xenograft models, repeated intravenous dosing of anti-CD19 or CD5 tLNPs carrying CAR mRNA led to CAR expression in of T cells, depending on construct design and dose, with higher levels observed using optimized CAR sequences. Tumor-engrafted mice treated with improved CD5tLNPs achieved robust CAR expression and tumor clearance. In cynomolgus monkeys, tLNPs induced dose-dependent CAR expression on circulating T cells and effective B cell depletion, demonstrating functional activity without tumor challenge. The tLNPs were based on ionizable lipid L829, modified with antibodies (e.g. anti-CD5, anti-CD8, anti-CD20) to enhance T-cell selectivity. This study highlights the translational potential of antibody-targeted LNPs for in vivo CAR T therapy [46].
Álvarez-Benedicto et al. developed spleen-targeted SORT LNPs incorporating anionic lipid 18:1 PA to deliver anti-CD19 CAR mRNA without antibody ligands. This strategy selectively transfected splenic T cells ~7% CAR+, extending survival in lymphoreplete B-cell lymphoma models while minimizing off-target lung and liver delivery [66].
Fernández Bimbo et al. developed NCtx, a DNA-based LNP system co-encapsulating CAR-encoding minicircle DNA and SB100x transposase mRNA, functionalized with anti-CD3 scFv and anti-CD7 nanobodies. A single 0.05 mg/kg intravenous dose in humanized PBMC or CD34+ xenograft models produced stable CAR-T cells with low PD-1 expression and a balanced memory/effector phenotype. Circulating CAR-T cells persisted for weeks, resulting in effective tumor control with low toxicity [67].
Metzloff et al. 2024 developed activating lipid nanoparticles (aLNPs) to streamline CAR T cell production by combining T cell activation and mRNA transfection into a single step. Engineered with CD3 and CD28 antibody fragments, aLNPs mimicked antigen-presenting cells and delivered CD19 CAR mRNA into primary human T cells. This eliminated the need for activating beads and reduced workflow time to 24 hours. aLNPs achieved >80% CAR T cell yield, efficient transfection, and generation of functional CAR T cells with robust proliferation, cytotoxicity, and activation phenotypes. In xenograft leukemia models, aLNP-derived CAR T cells demonstrated potent anti-tumor activity comparable to lentiviral approaches [68].
Overall, these studies highlight a range of innovative strategies for generating CAR-T cells directly in vivo. Approaches include antibody-mediated targeting of T cells (CD3, CD5, CD7), tuning ionizable lipids to favor extrahepatic delivery, stable genomic integration using transposases, organ-specific targeting, and the use of dual-gene payloads to modulate T-cell function. Preclinical evidence shows that antibody-conjugated LNPs can enhance transfection efficiency in circulating T cells while limiting off-target uptake by hepatocytes and other immune cells. Notably, these ligand-directed systems can also shape T-cell differentiation, with some studies reporting expansion of central memory–like populations when delivery is confined to specific subsets. Across different platforms, CAR-T transfection efficiencies reached up to ~20% in blood or spleen, depending on LNP composition, targeting strategy, and dose, underscoring the promise of LNPs as versatile, non-viral vehicles for in situ CAR-T engineering.
When benchmarked against ex vivo–manufactured CAR-T cells, LNP-generated cells have demonstrated competitive efficacy in murine models. Tumor clearance kinetics and durability of response have been comparable, although persistence of in vivo–engineered cells may be shorter in some contexts due to the transient nature of mRNA expression. Nevertheless, repeated dosing of LNPs has been explored as a strategy to extend therapeutic effect, capitalizing on the scalability and rapid manufacturing of nanoparticle formulations. Taken together, these preclinical findings provide a strong foundation for translation, establishing proof of feasibility across multiple antigens and tumor models.
Safety and immunogenicity considerations
Safety remains a central consideration in the development of LNP-based in vivo CAR-T therapies. Preclinical studies have highlighted several immunological and organspecific effects associated with these approaches [65-68]. Systemic T-cell activation is commonly observed following the administration of antibody-targeted LNPs, including anti-CD3 formulations, which induce activation markers, transient CD3 downregulation, and reversible depletion of circulating and splenic T cells. Importantly, T-cell populations typically recover within 24 hours, emphasizing the transient nature of these effects. Excessive activation, however, may contribute to systemic inflammation and organ toxicity, underscoring the need for careful dose optimization [44,46].
Cytokine Release Syndrome (CRS) and Immune effector Cell–Associated Neurotoxicity Syndrome (ICANS) remain potential risks, mirroring observations in conventional ex vivo CAR-T therapy [69] which arises from activated CAR-T cells releasing high levels of cytokines, including IL-6 and IFN-γ, which can precipitate systemic inflammation [70]. Preclinical evidence suggests that transient CAR expression via mRNA delivery and co-encapsulation of IL-6 shRNA can reduce CRS while maintaining antitumor efficacy [64] Antibody-directed targeting, organ-specific strategies such as Selective Organ Targeting (SORT) LNPs, have been shown to reduce hepatic and macrophage uptake while enhancing T-cell specificity. Even so, dose-dependent liver enzyme elevations have been observed at high LNP doses, and rare severe events, including hemophagocytic lymphohistiocytosis–like syndromes, underscore the need for cautious translational evaluation. Immunogenicity of the LNP carrier and payload represents an additional challenge [46]. Anti-PEG antibodies can accelerate LNP clearance [71] reducing efficacy and potentially inducing inflammatory responses, while repeated dosing of mRNA or plasmid DNA can trigger innate immune pathways, including STING, driving cytokine release [72].
Mitigation strategies have focused on rational LNP and payload design, targeted delivery, dosing optimization, and manufacturing control. Optimized ionizable lipids, helper lipids, and PEG lipids can reduce cytotoxicity and immunogenicity. Antibody-functionalized LNPs, Extrahepatic tropism, SORT LNPs, and CD47 decoration enhance T-cell specificity and minimize off-target effects. Co-delivery of functional modulators allows controlled immune activation, while transient CAR expression and local delivery reduce systemic toxicity [ 64]. Collectively, preclinical evidence indicates that LNP-mediated in vivo CAR-T generation carries manageable risks when carefully designed. Transient, tunable mRNA-based delivery combined with antibody or organ-targeted strategies minimizes off-target effects, mitigates systemic toxicity, and supports functional CAR-T activity, highlighting the translational potential of these approaches [44,46,64-68].
Early clinical translation and future directions
While in vivo CAR-T generation remains largely preclinical, early steps toward clinical translation are emerging. Several biotechnology companies have disclosed preclinical programs developing antibody-directed LNPs for T-cell engineering, and partnerships between academic centers and industry are accelerating progress. Capstan Therapeutics, for instance, is advancing targeted LNPs (tLNPs) capable of delivering CAR payloads directly to T cells in vivo, with AbbVie’s recent acquisition underscoring the translational momentum in this space [46]. Orna Therapeutics is pursuing a parallel strategy using circular RNA (oRNA®) formulated in LNPs to drive in vivo pan-CAR generation, supported by strategic alliances with Merck and Vertex [73]. Other emerging players such as ReNAgade and Orbital Therapeutics are also building broad RNA delivery platforms that include oncology applications. The scalability of LNP manufacturing, already validated in the global rollout of COVID-19 mRNA vaccines by Moderna and Pfizer/BioNTech, provides a unique advantage for clinical deployment, enabling rapid, large-scale production under GMP conditions [16,21]. The balance between transient versus durable expression remains an active area of investigation, with some groups pursuing hybrid strategies such as self-amplifying RNA to extend persistence while retaining the safety advantages of mRNA. To date, no clinical trial of LNP-based in vivo CAR-T has yet reported outcomes, but Investigational New Drug (IND)-enabling studies are underway. Key priorities include refining targeting specificity, validating safety in nonhuman primates, and determining optimal dosing strategies.
Discussion/conclusion
The potential clinical impact of this approach is profound. By eliminating ex vivo manipulation, LNP-based in vivo CAR-T could dramatically reduce cost, accelerate veinto-vein time, and expand access globally, particularly in regions where current therapies remain excessively expensive or logistically infeasible. If safety and efficacy are confirmed in human trials, this strategy may redefine the landscape of cellular immunotherapy, transitioning CAR-T therapy from a personalized, resource-intensive procedure to an off-the-shelf modality deliverable at scale.
References
- Zhang X, Zhu L, Zhang H, Chen S, Xiao Y. CAR-T cell therapy in hematological malignancies: current opportunities and challenges. Front Immunol. 2022; 13: 927153.
- S S KD, Joga R, Srivastava S, Nagpal K, Dhamija I, Grover P, et al. Regulatory landscape and challenges in CAR-T cell therapy development in the US, EU, Japan, and India. Eur J Pharm Biopharm. 2024; 201: 114361.
- Goyco Vera D, Waghela H, Nuh M, Pan J, Lulla P. Approved CAR-T therapies have reproducible efficacy and safety in clinical practice. Hum Vaccin Immunother. 2024; 20: 2378543.
- Mallapaty S. Cutting-edge CAR-T cancer therapy is now made in India at one-tenth the cost. Nature. 2024; 627: 709-10.
- Narula G, Keerthivasagam S, Jain H, Punatar S, Chichra A, Dhamne C, et al. Correction to: Novel humanized CD19-CAR-T (Now talicabtagene autoleucel, Tali-celTM) cells in relapsed/refractory pediatric B-acute lymphoblastic leukemia an open-label single-arm phase-I/Ib study. Blood Cancer J. 2025; 15.
- Jain H, Karulkar A, Kalra D, Ravikumar S, Shah S, Firfiray A, et al. Talicabtagene autoleucel for relapsed or refractory B-cell malignancies: results from an open-label, multicentre, phase 1/2 study. Lancet Haematol. 2025; 12: e282-93.
- Abou-El-Enein M, Elsallab M, Feldman SA, Fesnak AD, Heslop HE, Marks P, et al. Scalable manufacturing of CAR T cells for cancer immunotherapy. Blood Cancer Discov. 2021; 2: 408.
- Gee AP. GMP CAR-T cell production. Best Pract Res Clin Haematol. 2018; 31: 126-34.
- Levine BL, Miskin J, Wonnacott K, Keir C. Global manufacturing of CAR T cell therapy. Mol Ther Methods Clin Dev. 2016; 4: 92.
- Bauer G, Fury B. Challenges of translating a cell therapy to GMP. Int Rev Neurobiol. 2022; 166: 207-34.
- Ceballos C, Viguria MC, Panizo C, Rodríguez-Madoz JR, Prósper F. Advances and challenges in CAR-T cell therapy: from early chimeric antigen receptors to future frontiers in oncology. Front Hematol. 2023; 2: 1217775.
- Sureda A, Adam SE, Yang S, Griffin E, Baker J, Johnston K, et al. Logistical challenges of CAR T-cell therapy in non-Hodgkin lymphoma: a survey of healthcare professionals. Future Oncol. 2024; 20: 2855-68.
- Dias J, Garcia J, Agliardi G, Roddie C. CAR-T cell manufacturing landscape—lessons from the past decade and considerations for early clinical development. Mol Ther Methods Clin Dev. 2024; 32: 101250.
- Ayala Ceja M, Khericha M, Harris CM, Puig-Saus C, Chen YY. CAR-T cell manufacturing: major process parameters and next-generation strategies. J Exp Med. 2024; 221.
- Bhaskar ST, Dholaria BR, Sengsayadeth SM, Savani BN, Oluwole OO. Role of bridging therapy during chimeric antigen receptor T cell therapy. EJHaem. 2021; 3: 39.
- Meng S, Hara T, Miura Y, Arao Y, Saito Y, Inoue K, et al. In vivo engineered CAR-T cell therapy: lessons built from COVID-19 mRNA vaccines. Int J Mol Sci. 2025; 26: 3119.
- Moretti A, Ponzo M, Nicolette CA, Tcherepanova IY, Biondi A, Magnani CF. The past, present, and future of non-viral CAR T cells. Front Immunol. 2022; 13.
- Karlsson J, Rhodes KR, Green JJ, Tzeng SY. Poly(beta-amino ester) s as gene delivery vehicles: challenges and opportunities. Expert Opin Drug Deliv. 2020; 17: 1395.
- Grewal AK, Salar RK. Chitosan nanoparticle delivery systems: an effective approach to enhancing efficacy and safety of anticancer drugs. Nano TransMed. 2024; 3: 100040.
- Urits I, Swanson D, Swett MC, Patel A, Berardino K, Amgalan A, et al. A review of Patisiran (ONPATTRO) for the treatment of polyneuropathy in people with hereditary transthyretin amyloidosis. Neurol Ther. 2020; 9: 301.
- Lewis LM, Badkar AV, Cirelli D, Combs R, Lerch TF. The race to develop the Pfizer-BioNTech COVID-19 vaccine: from the pharmaceutical scientists’ perspective. J Pharm Sci. 2023; 112: 640-7.
- Habrant D, Peuziat P, Colombani T, Dallet L, Gehin J, Goudeau E, et al. Design of ionizable lipids to overcome the limiting step of endosomal escape: application in the intracellular delivery of mRNA, DNA, and siRNA. J Med Chem. 2016; 59: 3046-62.
- Swaminathan G, Thoryk EA, Cox KS, Smith JS, Wolf JJ, Gindy ME, et al. A tetravalent sub-unit dengue vaccine formulated with ionizable cationic lipid nanoparticle induces significant immune responses in rodents and non-human primates. Sci Rep. 2016; 6.
- Buck J, Grossen P, Cullis PR, Huwyler J, Witzigmann D. Lipid-based DNA therapeutics: hallmarks of non-viral gene delivery. ACS Nano. 2019; 13: 3754-82.
- Wang X, Liu S, Sun Y, Yu X, Lee SM, Cheng Q, et al. Preparation of selective organ-targeting lipid nanoparticles using multiple technical methods for tissue-specific mRNA delivery. Nat Protoc. 2022; 18: 265.
- Strelkova Petersen DM, Chaudhary N, Arral ML, Weiss RM, Whitehead KA. The mixing method used to formulate lipid nanoparticles affects mRNA delivery efficacy and organ tropism. Eur J Pharm Biopharm. 2023; 192: 126-35.
- Eshhar Z, Waks T, Gkoss G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the γ or ζ subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA. 1993; 90: 720-4.
- Dwivedi A, Karulkar A, Ghosh S, Srinivasan S, Kumbhar BV, Jaiswal AK, et al. Robust antitumor activity and low cytokine production by novel humanized anti-CD19 CAR T cells. Mol Cancer Ther. 2021; 20: 846-58.
- Jayaraman J, Mellody MP, Hou AJ, Desai RP, Fung AW, Pham AHT, et al. CAR-T design: elements and their synergistic function. EBioMedicine. 2020; 58: 102931.
- Tang L, Pan S, Wei X, Xu X, Wei Q. Arming CAR-T cells with cytokines and more: innovations in the fourth-generation CAR-T development. Mol Ther. 2023; 31: 3146-62.
- Gomes-Silva D, Ramos CA. Cancer immunotherapy using CAR-T cells: From the research bench to the assembly line. Biotechnol J. 2018; 13: 2.
- Seton-Rogers S. Programming T cells in situ. Nat Rev Cancer. 2017; 17: 372–72.
- Zhang W, Huang X. In vivo gene editing and in situ generation of chimeric antigen receptor cells for next-generation cancer immunotherapy. J Hematol Oncol. 2024; 17: 1–23.
- Frank AM, Braun AH, Scheib L, Agarwal S, Schneider IC, Fusil F, et al. Combining T-cell-specific activation and in vivo gene delivery through CD3-targeted lentiviral vectors. Blood Adv. 2020; 4: 5702–15.
- Michels KR, Sheih A, Hernandez SA, Brandes AH, Parrilla D, Irwin B, et al. Preclinical proof of concept for VivoVec, a lentiviral-based platform for in vivo CAR T-cell engineering. J Immunother Cancer. 2023; 11.
- Nicolai CJ, Parker MH, Qin J, Tang W, Ulrich-Lewis JT, Gottschalk RJ, et al. In vivo CAR T-cell generation in nonhuman primates using lentiviral vectors displaying a multidomain fusion ligand. Blood. 2024; 144: 977–87.
- McCarty DM, Young SM, Samulski RJ. Integration of adeno-associated virus and recombinant AAV vectors. Annu Rev Genet. 2004; 38: 819–45.
- Ciuffi A. Mechanisms governing lentivirus integration site selection. Curr Gene Ther. 2008; 8: 419–29.
- Bulaklak K, Gersbach CA. The once and future gene therapy. Nat Commun. 2020; 11: 5820.
- Bulcha JT, Wang Y, Ma H, Tai PWL, Gao G. Viral vector platforms within the gene therapy landscape. Signal Transduct Target Ther. 2021; 6: 1–24.
- Zhao Z, Anselmo AC, Mitragotri S. Viral vector-based gene therapies in the clinic. Bioeng Transl Med. 2021; 7: e10258.
- Taghdiri M, Mussolino C. Viral and non-viral systems to deliver gene therapeutics to clinical targets. Int J Mol Sci. 2024; 25: 7333.
- Billingsley MM, Singh N, Ravikumar P, Zhang R, June CH, Mitchell MJ. Ionizable lipid nanoparticle-mediated mRNA delivery for human CAR T cell engineering. Nano Lett. 2020; 20: 1578–89.
- Kheirolomoom A, Kare AJ, Ingham ES, Paulmurugan R, Robinson ER, Baikoghli M, et al. In situ T-cell transfection by anti-CD3-conjugated lipid nanoparticles leads to T-cell activation, migration, and phenotypic shift. Biomaterials. 2021; 281: 121339.
- Metzloff AE, Padilla MS, Gong N, Billingsley MM, Han X, Merolle M, et al. Antigen presenting cell mimetic lipid nanoparticles for rapid mRNA CAR T cell cancer immunotherapy. Adv Mater. 2024; 36: 2313226.
- Hunter TL, Bao Y, Zhang Y, Matsuda D, Riener R, Wang A, et al. In vivo CAR T cell generation to treat cancer and autoimmune disease. Science. 2025; 388: 1311–7.
- Zeng Y, Escalona-Rayo O, Knol R, Kros A, Slütter B. Lipid nanoparticle-based mRNA candidates elicit potent T cell responses. Biomater Sci. 2022; 11: 964–74.
- Alshehry Y, Liu X, Li W, Wang Q, Cole J, Zhu G. Lipid nanoparticles for mRNA delivery in cancer immunotherapy. AAPS J. 2025; 27: 3.
- Koltover I, Salditt T, Rädler JO, Safinya CR. An inverted hexagonal phase of cationic liposome-DNA complexes related to DNA release and delivery. Science. 1998; 281: 78–81.
- Ye Z, Chen J, Zhao X, Li Y, Harmon J, Huang C, et al. In vitro engineering chimeric antigen receptor macrophages and T cells by lipid nanoparticle-mediated mRNA delivery. ACS Biomater Sci Eng. 2022; 8: 722–33.
- Kulkarni JA, Witzigmann D, Leung J, Tam YYC, Cullis PR. On the role of helper lipids in lipid nanoparticle formulations of siRNA. Nanoscale. 2019; 11: 21733–9.
- Zhang R, El-Mayta R, Murdoch TJ, Warzecha CC, Billingsley MM, Shepherd SJ, et al. Helper lipid structure influences protein adsorption and delivery of lipid nanoparticles to spleen and liver. Biomater Sci. 2021; 9: 1449.
- Zhang L, Seow BYL, Bae KH, Zhang Y, Liao KC, Wan Y, et al. Role of PEGylated lipid in lipid nanoparticle formulation for in vitro and in vivo delivery of mRNA vaccines. J Control Release. 2025; 380: 108–24.
- Waggoner LE, Miyasaki KF, Kwon EJ. Analysis of PEG-lipid anchor length on lipid nanoparticle pharmacokinetics and activity in a mouse model of traumatic brain injury. Biomater Sci. 2023; 11: 4238.
- Berger M, Degey M, Leblond Chain J, Maquoi E, Evrard B, Lechanteur A, et al. Effect of PEG anchor and serum on lipid nanoparticles: Development of a nanoparticles tracking method. Pharmaceutics. 2023; 15: 2.
- Jürgens DC, Müller JT, Nguyen A, Merkel OM. Tailoring lipid nanoparticles for T-cell targeting in allergic asthma: Insights into efficacy and specificity. Eur J Pharm Biopharm. 2024; 198: 114242.
- Patel S, Ashwanikumar N, Robinson E, Xia Y, Mihai C, Griffith JP, et al. Naturally-occurring cholesterol analogues in lipid nanoparticles induce polymorphic shape and enhance intracellular delivery of mRNA. Nat Commun. 2020; 11: 983.
- Kawaguchi M, Noda M, Ono A, Kamiya M, Matsumoto M, Tsurumaru M, et al. Effect of cholesterol content of lipid composition in mRNA-LNPs on the protein expression in the injected site and liver after local administration in mice. J Pharm Sci. 2022; 112: 1401.
- Patel SK, Billingsley MM, Frazee C, Han X, Swingle KL, Qin J, et al. Hydroxycholesterol substitution in ionizable lipid nanoparticles for mRNA delivery to T cells. J Control Release. 2022; 347: 521– 32.
- Lokras AG, Baghel SS, Jensen RF, Thakur A, Franzyk H, Thofte O, et al. Cholesterol in mRNA-lipid nanoparticles can be replaced with the synthetic mycobacterial monomycoloyl glycerol analogue MMG-1. Adv Funct Mater. 2025; e05627.
- Ginaldi L, Farahat N, Matures E, De Martinis M, Morilla R, Catovsky D. Differential expression of T cell antigens in normal peripheral blood lymphocytes: a quantitative analysis by flow cytometry. J Clin Pathol. 1996; 49: 539.
- Van De Parre TJL, Martinet W, Schrijvers DM, Herman AG, De Meyer GRY. mRNA but not plasmid DNA is efficiently transfected in murine J774A.1 macrophages. Biochem Biophys Res Commun. 2005; 327: 356–60.
- Youn H, Chung JK. Modified mRNA as an alternative to plasmid DNA for transcript replacement and vaccination therapy. Expert Opin Biol Ther. 2015; 15: 1337.
- Zhou J, Sun L, Jia Y, Wang Z, Luo T, Tan J, et al. Lipid nanoparticles produce chimeric antigen receptor T cells with interleukin-6 knockdown in vivo. J Control Release. 2022; 350: 298–307.
- Billingsley MM, Gong N, Mukalel AJ, Thatte AS, El-Mayta R, Patel SK, et al. In vivo mRNA CAR T cell engineering via targeted ionizable lipid nanoparticles with extrahepatic tropism. Small. 2024; 20.
- Álvarez-Benedicto E, Tian Z, Chatterjee S, Orlando D, Kim M, Guerrero ED, et al. Spleen SORT LNP generated in situ CAR T cells extend survival in a mouse model of lymphoreplete B cell lymphoma. Angew Chem Int Ed. 2023; 62.
- Bimbo JF, van Diest E, Murphy DE, Ashoti A, Evers MJW, Narayanavari SA, et al. T cell-specific non-viral DNA delivery and in vivo CAR-T generation using targeted lipid nanoparticles. J Immunother Cancer. 2025; 13: e011759.
- Metzloff AE, Padilla MS, Gong N, Billingsley MM, Han X, Merolle M, et al. Antigen presenting cell mimetic lipid nanoparticles for rapid mRNA CAR T cell cancer immunotherapy. Adv Mater. 2024; 36: e2313226.
- Epperly R, Giordani VM, Mikkilineni L, Shah NN. Early and late toxicities of CAR T-cells. Hematol Oncol Clin North Am. 2023; 37: 1169.
- Sterner RC, Sterner RM. Immune effector cell associated neurotoxicity syndrome in chimeric antigen receptor-T cell therapy. Front Immunol. 2022; 13: 879608.
- Wang H, Wang Y, Yuan C, Xu X, Zhou W, Huang Y, et al. Polyethylene glycol-associated immune responses triggered by clinically relevant lipid nanoparticles in rats. NPJ Vaccines. 2023; 8: 1–13.
- Verbeke R, Hogan MJ, Loré K, Pardi N. Innate immune mechanisms of mRNA vaccines. Immunity. 2022; 55: 1993.
- Orna Therapeutics. Our pipeline. 2025.