Journal of
Clinical & Medical Surgery
Clinical & Medical Surgery
www.jclinmedsurgery.com
ISSN 2833-5465
Open Access
Volume 3
Open Access
Volume 3
*Corresponding Author: GK Parvathidevi
Sri Siddhartha medical college & research center, Tumkur-572107, Karnataka, India.
Email: drgkparvathidevi27@gmail.com
Article Info
Received: May 22, 2023
Accepted: Jun 23, 2023
Published: Jun 30, 2023
Archived: www.jclinmedsurgery.com
Copyright: ©Parvathidevi GK (2023).
Citation: Parvathidevi GK. Endogenous Ochronosis: A Rare Case Report. J Clin Med Surgery. 2023; 3(1): 1107.
Introduction
The term “OCHRONOSIS” was first described by Virchow in 1866 as a brownish-yellow pigment that gets deposited in the connective tissue of various organs. Ochronosis can be endogenous or exogenous in origin. Endogenous ochronosis is caused by deficiency of ochronosis is caused by deficiency of the enzyme homogentisic acid oxidase which causes deposition of ochronotic pigmented. In contrast, exogenous ochronosis presents as gray-brown or blue-black macules, hyperchromic, pinpoint papules in photo-exposed regions in a symmetrical pattern and occurs secondary to the topical application of hydroquinone, phenol, resorcinol or even by oral administration of antimalarials.
Endogenous ochronosis is also called Alkaptonuria is a rare metabolic autosomal recessive disease which affects 1:1000000 people. Alkaptonuria though present at birth, but usually manifested in adults, starting after the age of thirty years.
Case presentation
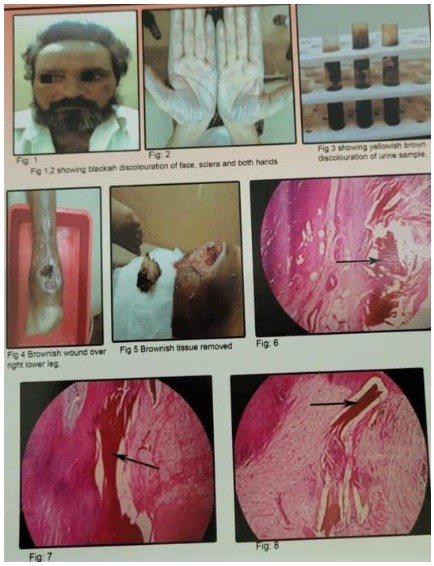
We report a case of endogenous ochronosis in 45 year old man who presented with a blackish wound over the lower posterior part of right leg and blackish discoloration of face and both palms (Figures 1,2). With clinical examination and urine examination (Figure 3), a provisional diagnosis of ochronosis was made. We received a specimen of right tendoachilles (Figures 2,5). Microscopically revealed fibrocollagenous tendinous tissue showing homogenous and granular deposits of brownish pigment (Figures 6,7,8) [1-3].
Discussion
Endogenous ochronosis is a rare metabolic autosomal recessive disorder with tolat deficiency of homogentisic acid oxidase enzyme. The homogentisic acid is part of metabolic pathway of phenylalanine and tyrosine. The deficiency of the enzyme that metabolises it (homogentisic acid oxidase) leads to its accumulation, that will be polymerised in a melanin-like pigment that presents high affinity for connective tissue, espically cartilage, resulting in an ocher color (for this reason it carries the name of ochronosis).
In children its main symptom is darkening of urine after a long period of rest or in contact with the environmental or alkali air, as well as blackened spots in babies diapers, whereas in adults, after the fourth decade in life, the main manifestation is osteoarthritis, followed by changes in eyes, ears, skin and in genitourinary, cardiovascular and musculoskeletal system.
Its excess in deposition mainly in cartilaginous tissue, mucous, skin, bone surface and internal cardiac structures, as well as excreted in biological solutions (urine, sweat and semen). The main complications of alkaptonuria are vascular calcifications and osteoarthritis, mainly in the joints of the cervical spine, besides dark pigmentation of skin, cartilage, sclera and other connective tissues. The deposition of pigment is observed in joints that suffer great pressure, like in the lumbar and large articulations. This occurs because the ochronosis pigment has high affinity for the collagen fibers of articulations.
The diagnosis is done through the clinical history and from changes in the coloration of urine in environmental or alkali air and confirmed by dosage of homogentisic acid in urine, which is gold standard. Currently there is no effective treatment for alkaptonuria and management is symptomatic. Arthropathy is usually treated with NSAIDs; however, in advanced cases, join replacement surgery provides significant relief from symptoms.
Conclusion
Endogenous ochronosis is a rare metabolic autosomal recessive disorder with total deficiency of homogentisic acid oxidase enzyme that leads to the deposition of a melanin-like pigment in various tissues.
References
- Sharma S, Rao R. Exogenous ochronosis masquerading refractory melasma. Our dermatol online. 2014; 5: 398-400.
- Craide FH, da Fonseca JSBM, Mariano PC, Fernandez NM, de Castro CGC, et al. Alkaptonuria-case report. An Bras dermatol. 2014; 89: 799-801.
- Babanagare SV, Deshmuk SD, Khadilkar MS, Patil AA. Ochronosis: A report of three cases and review of the literature. Indian J Pathol Microbiol. 2011; 54: 626-628.